Studi e terapie innovative
USO DELL’ INSULINA NELLA CURA DEL GLAUCOMA AD ANGOLO APERTO E DI ALTRE PATOLOGIE A CARICO DELLA RETINA
Il glaucoma, una delle principali cause di cecità irreversibile in tutto il mondo; è caratterizzato da una perdita permanente delle cellule gangliari della retina, un gruppo di neuroni del sistema nervoso centrale che trasmettono le informazioni visive dalla retina al cervello attraverso terminazioni nervose dette assoni.
Clinicamente, il danno assonale in questa patologia provoca una perdita del campo visivo e può portare alla cecità. Attualmente, la riduzione della pressione oculare rimane l’unico obiettivo delle terapie comprovate per il glaucoma. Tuttavia, molti pazienti continuano a perdere la vista anche quando vengono attuati interventi standard.
I dendriti sono tipicamente ramificazioni neuronali che determinano come questi ultimi ricevono e integrano le informazioni. La retrazione dei dendriti e la rottura delle sinapsi sono segni precoci di diversi disturbi neuro/degenerativi. I neuroni hanno una capacità estremamente limitata di rigenerarsi dopo una lesione.
Recentemente l’insufficiente segnalazione dell’insulina è stata implicata in malattie caratterizzate da patologia dendritica, in particolare il morbo di Alzheimer e il glaucoma. l’insulina infatti attraversa la barriera emato-encefalica e influenza numerosi processi cerebrali.
Studi recenti hanno dimostrato che la somministrazione di insulina dopo la lesione del nervo ottico ha provocato una robusta ricrescita dendritica, la sopravvivenza delle cellule gangliari della retina e il salvataggio delle risposte retiniche, fornendo la prima prova di una rigenerazione dendritica riuscita nei neuroni dei mammiferi. Una ricerca [1] convalida la terapia insulinica come un potente farmaco per ripristinare la funzione dendritica nel glaucoma, formando la base per l’utilizzo dell’insulina come trattamento del glaucoma negli esseri umani.
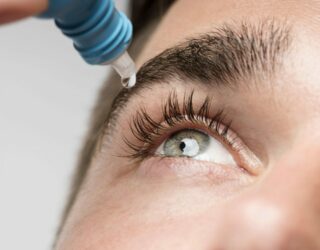
Attualmente, l’insulina è largamente utilizzata nella cura del diabete. Gli eventi avversi comprendono ipoglicemia, ipokaliemia, lipodistrofia, allergie, aumento di peso, edema periferico e interazioni farmacologiche. L’uso sperimentale di insulina topica oculare è stato testato in piccole coorti di individui sani e pazienti diabetici, senza riportare eventi avversi significativi. Tuttavia, questi protocolli variavano nella posologia dell’insulina e gli eventi avversi sono stati solo accennati brevemente, indicando la necessità di caratterizzare meglio il profilo di sicurezza di tale uso off-label dell’insulina prima della sua applicazione come trattamento neuroprotettivo e rigenerativo per il glaucoma.
In questo studio, i ricercatori ipotizzano che l’insulina oculare topica (fino a 500 U/ml) una volta al giorno sia sicura nei pazienti con glaucoma ad angolo aperto.
Natura sperimentale del farmaco: trattamento
Secondo lo studio a cui stiamo riferendoci, applicazione topica di insulina con concentrazioni di 100 U/ml [2] e 500 U/ml [3] una volta al giorno agli occhi con diagnosi di glaucoma ad angolo aperto. (entrambi i prodotti di insulina sono approvati da Health Canada per uso sottocutaneo ed endovenoso per il trattamento del diabete mellito). La via di somministrazione proposta e l’indicazione dell’uso di insulina in questo studio sono di natura off-label, ma vuole dimostrare che l-uso dell’insulina oculare topica (fino a 500 U/ml) una volta al giorno è sicura nei pazienti con glaucoma ad angolo aperto, questo documentando e segnalando eventuali eventi avversi oculari e/o sistemici associati a colliri insulinici topici.
Bibliografia
[1] – Sicurezza delle gocce di insulina per uso topico per il glaucoma ad angolo aperto – 4 aprile 2023 – Centre hospitalier de l’Université de Montréal (CHUM) Registro degli studi clinici negli Stati Uniti – Sperimentazione clinica NCT04118920
[2] – Humulin R U-100, Eli Lilly Canada, St-Laurent, Quebec, Canada
[3] – Entuzity, Eli Lilly Canada, St-Laurent, Quebec, Canada

 Nel trial clinico citato, sono stati coinvolti 31 pazienti con Stargardt trattati con 20 milligrammi al giorno di zafferano (Repron, brevetto internazionale) in compresse. I pazienti hanno assunto lo zafferano per sei mesi e poi una sostanza placebo per i successivi sei.
Nel trial clinico citato, sono stati coinvolti 31 pazienti con Stargardt trattati con 20 milligrammi al giorno di zafferano (Repron, brevetto internazionale) in compresse. I pazienti hanno assunto lo zafferano per sei mesi e poi una sostanza placebo per i successivi sei. In quanto carotenoide, il licopene è contenuto soprattutto in alcuni alimenti del regno vegetale. Se consideriamo il contenuto di licopene nei vari cibi, il pomodoro è sicuramente l’alimento principe (ne contiene da 3 a 40 mg per kg di prodotto fresco). Altre fonti minori sono rappresentate da vegetali come pompelmo rosa, arance rosse, carote, albicocche e cocomeri.
In quanto carotenoide, il licopene è contenuto soprattutto in alcuni alimenti del regno vegetale. Se consideriamo il contenuto di licopene nei vari cibi, il pomodoro è sicuramente l’alimento principe (ne contiene da 3 a 40 mg per kg di prodotto fresco). Altre fonti minori sono rappresentate da vegetali come pompelmo rosa, arance rosse, carote, albicocche e cocomeri.